
COVID-19患者的已知风险因素主要是先前存在的疾病,例如老年、高血压、肥胖、糖尿病等,通过这些因素预测疾病严重程度是非常不准确的,而更精确的疾病严重程度分类可能会产生重要的临床结果。
到目前为止,绝大多数对COVID-19患者的健康状况或严重程度进行分类的尝试都依赖于TCR数据。BCR测序的使用被认为比TCR更困难,因为体细胞超突变(SHM)和CDR3区的多样性更高。然而,在某些情况下,BCR数据可能比TCR更具信息性,因为BCR会经历亲和力成熟以适应每种病原体。
2023年4月发表于Frontiers in Immunology的一篇文章报告了基于机器学习(ML)的BCR测序成功地将SARS-CoV2感染者与非感染者进行了分类,并确定疾病的严重程度,驱动这种分类的特征基于体细胞超突变模式。这些结果可用于建立和调整针对COVID-19的治疗策略,为未来的流行病学挑战提供新思路。
参考文献:Safra M, Tamari Z, Polak P, et al . Altered somatic hypermutation patterns in COVID-19 patients classifies disease severity. Front Immunol. 2023 Apr 19;14:1031914.
研究方法:样本采集(39名COVID-19轻度患者、12名严重感染患者和28个对照样本,采集PBMC)→bulk测序和单细胞测序→数据处理和统计→生成SHM模型→ML算法的训练和估计 →使用AA频率进行COVID-19分类→单细胞数据分析

BCR基因的使用无法对SARS-CoV2感染进行分类
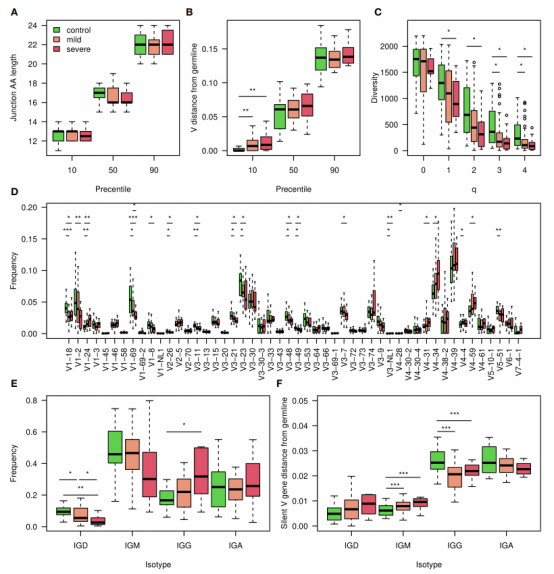
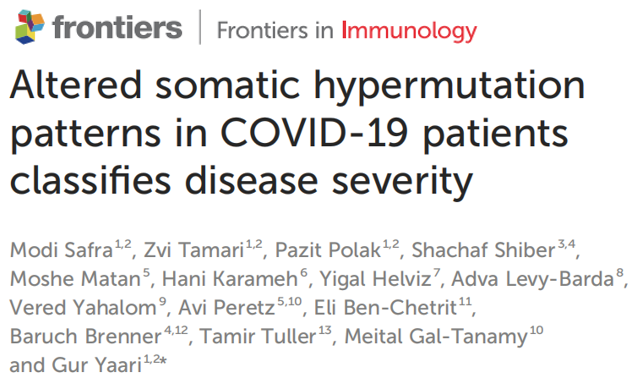
图1.COVID-19 BCR重链的特征
与对照组相比,COVID-19患者的CDR3氨基酸(AA)长度无显著性差异(图1A),V基因突变分布仅略有增加(图1B),BCR克隆多样性明显偏低(图1C)。COVID-19患者的V基因使用率显著减少,但IGHV4-34,IGHV4-39和IGHV4-59的使用率增加,且重度患者高于轻度患者(图1D)。表明针对SARS-CoV2的抗体主要由这些基因组成。
作者根据V基因或V & J基因使用情况或V&J基因使用情况以及CDR3 AA中85%的相似性构建ML分类器。然而,这些模型的准确率低于70%,表明V或V&J基因的使用对SARS-CoV2感染应答的影响较小。
比较不同组之间的isotype频率发现,SARS-CoV2感染后IGD和IGM的频率降低,IGG的频率升高,而IGA的频率保持不变(图1E)。每个isotype的沉默突变(silent mutability)频率比较:感染后IGG和IGA的突变性降低,IGD和IGM的突变性增加,重症患者更高(图1F)。

BCR V基因AA组成成功对SARS-CoV2感染进行分类,并可能揭示病毒抗体的重要特征
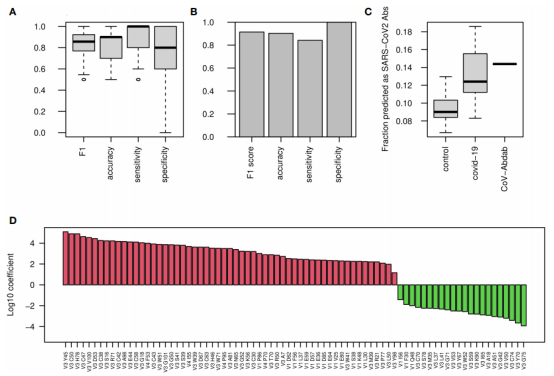
图2.使用所有V基因位置的AA频率进行COVID-19分类
作者生成了一个包含10300列的表,为5个最常用的V基因家族(IGHV1-5)沿103个V基因位置(根据IMGT编号对齐)计算AA频率。使用这种方法获得了超过0.85的高 F1分数,以及相似水平的准确性、灵敏度和特异性(图 2A). 外部测试组的F1分数也高于0.85(图2B).作者提取了该算法使用的系数,对应于每个AA频率对疾病分类的贡献(图2D).另外作者计算了来自CoV-AbDab数据库中5000多种已知的抗SARS-CoV2抗体的这些分数,已知抗体的评分高于对照组以及大多数COVID-19感染库的评分(图2C),表明这些系数不仅对库水平有意义,而且对单个BCR序列也有意义。

COVID-19患者的类别转换B细胞的突变偏倚
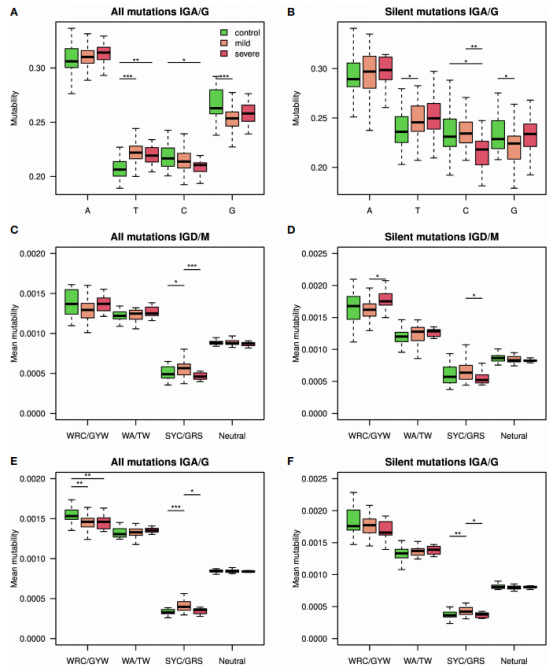
图3.SHM单碱基突变、5-mers热点和冷点的沉默和替换突变
如图3A所示,COVID-19患者在胞嘧啶和鸟嘌呤(C和G)时的平均相对突变性降低,在腺嘌呤和胸腺嘧啶(A和T)的平均相对突变性增加。当只考虑沉默突变时,也得到了相同的结果(图3B)。
作者建立了一个基于沉默突变和替换突变的5-mer突变模型。这种模型结合了SHM和抗原驱动选择的影响。将5-mers分为发生在两个热点(WRC/GYW和WA/TW)、冷点(SYC/GRS)和所有其他中性位点,图3C、E显示了IGD/IGM和IGA/ IGG的水平。不同组间最显著的变化是COVID-19患者IGA/IGG中WRC/GYW位点减少,SYC/GRS增加,这种增加在严重感染的患者中没有出现。
为了了解这些模式是来自SHM还是来自抗原驱动的选择,作者建立了另一个模型,只考虑了沉默突变。图3D,F显示了相同序列基序的突变性分数。与图3C,E中观察到的模式相似,表明两组之间的变化是由于SHM特征的改变造成的。

Silent SHM模式对SARS-CoV2感染和严重程度进行了分类
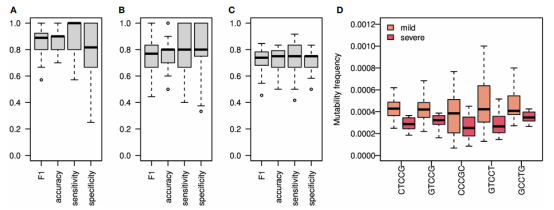
图4.SHM重链可以对SARS-CoV2感染和严重程度进行分类
为了估计SHM模式的变化与SARS-CoV2感染之间的联系水平,作者建立了两个模型,一个使用了所有突变(图4A),另一个只使用了沉默突变(图4B)。考虑所有突变时,获得的F1评分和准确性、敏感性和特异性值均很高;只考虑沉默突变时,得到的F1评分和准确性的结果略低。这些结果印证了基因库之间的差异主要来自于SHM本身,而不是来自于抗原驱动的选择。
由于冷点基序的突变性在重症和轻度患者之间变化最大,作者仅使用该冷点的突变性评分建立了一个模型。在严重程度分类中,获得的F1评分和准确性约为0.75(图4C)。与重症患者相比,所有轻症患者的非零系数模式都有更高的突变频率(图4D)。

已知的SARS-CoV2抗体在COVID-19患者的浆母细胞中富集
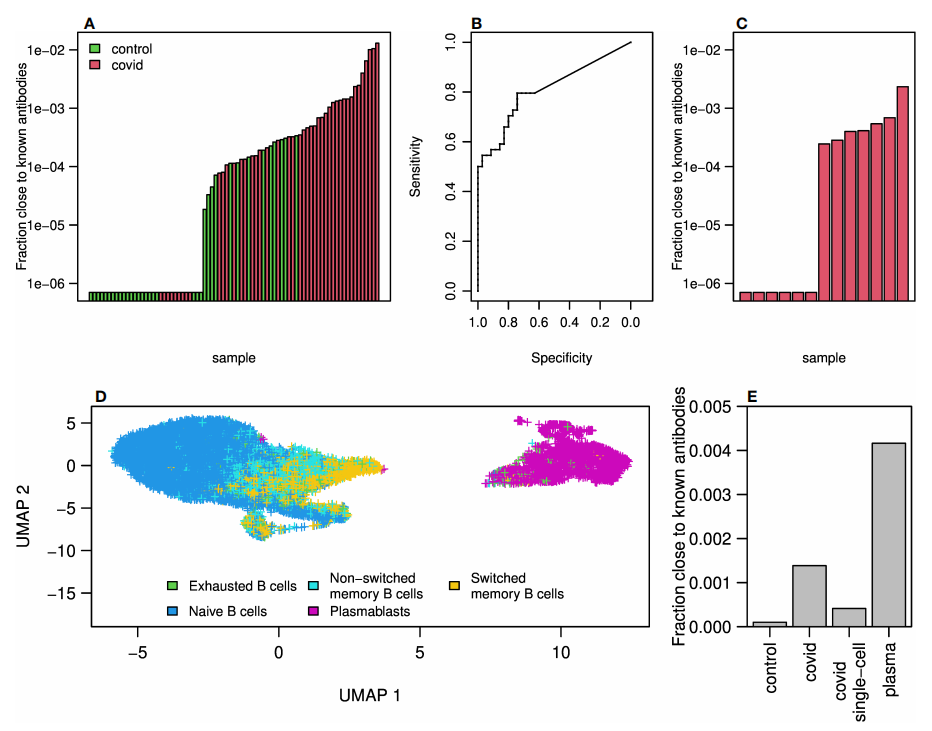
图5.测序中的抗体克隆接近CoV-AbDab数据库中已知的COVID-19抗体
作者计算并总结了测序得到的抗体库与已公布的已知SARS-CoV2抗体相似的序列的频率。COVID-19患者中与已知抗体相似的频率高于对照组(图5A)。利用与已知COVID-19克隆相似的频率之和,我们在曲目分类中达到了70%以上的准确率,AUC为0.81(图5B)。
为了进一步探索与已知抗体的相似性,作者对另外13例轻度COVID-19患者的血液样本进行了10X基因组学单细胞测序。在其中7个抗体库中发现了与已知抗体相似的抗体序列(图5C),与批量测序队列相比,这些频率总体上较低。这可能是由于测序方法的差异,或者是因为在单细胞队列中,患者的平均诊断时间比整体队列要晚,因此可能有较低的SARS-CoV2特异性抗体水平。
图5D是根据单细胞表达谱对细胞类型分类的二维UMAP简化图,显示了一组独特的浆母细胞群(图中的紫色)。
已知的SARS-CoV2抗体在批量测序患者组、批量对照组、单细胞未细胞分类时和单细胞浆母细胞中的频率比较如图5E所示,已知抗体在COVID-19患者浆母细胞中的频率最高,这表明了对SARS-CoV2的典型反应。

验证
为了验证上述基于SHM突变模式的ML算法是否特定于 COVID-19,作者将算法应用于450份样本,样本来自乙型肝炎病毒感染、乙型肝炎病毒和流感疫苗接种以及几种自身免疫性疾病患者。这些样本中有94%被归类为健康,这表明这套算法不会将针对新病原体的免疫反应归类为COVID-19,它是特定于 COVID-19的。
沿V基因的AA频率模式是相对良好地对COVID-19进行分类的标志物,SHM突变模式可以实现对COVID-19严重程度分类,确定了与严重程度相关的SHM特异性模式,以及SARS-CoV2抗体中重要的AA组成。


杭州艾沐蒽生物科技有限公司成立于2016年,是国内前沿的专注于免疫基因组学技术的国家高新技术企业。创始人团队来自美国芝加哥大学,在2010年开始使用免疫组高通量测序技术开展各种疾病相关研究,于2016年通过自主研发,全国首家推出NGS-MRD血液肿瘤微小残留病(MRD)检测Seq-MRD®,并授权泛生子(纳斯达克代码:GTH)使用。同时,公司拥有Immun-Traq®肿瘤治疗伴随诊断、Immun-Cheq® |T细胞免疫测评以及ImmuHub®免疫组测序科研服务产品,并布局有基于AI机器学习算法的T-classifier®疾病早筛、单细胞测序、TCR-T药物开发等平台管线。公司构建几十项发明专利和软件著作权为核心的自主知识产权体系,为医院临床、生命科学研究、新药开发等提供解决方案和产品。
艾沐蒽专注于通过解码适应性免疫系统来改变疾病的诊断和治疗,并致力于推进免疫驱动医学领域发展。

